
(1)
Germán Rodas
![]()
![]()
![]()
1 Médico; Egresado del Postgrado de Oncología Clínica, Universidad San Francisco de Quito, Hospital Carlos Andrade Marín.
Correspondencia:
Dr. Germán Rodas
E-mail: germanrodas@hotmail.com
![]()
Recibido: 06 – Abril – 2013
Aceptado: 21 – Junio – 2013
![]()
Palabras clave: Neoplasias del tejido nervioso; Neurofibroma; Neurofibromatosis;
Reporte de caso.
Forma de citar este artículo:
Rodas G. Neurofibroma con alto grado de atipia en una paciente con neurofibromatosis tipo 1.
Rev Med Vozandes 2013; 24: 79 – 82.
La neurofibromatosis de tipo 1 tiene una prevalencia de un caso en cada 2500 a 3000 personas. Es una enfermedad autosómica dominante que presenta básicamente ausencia de una proteína llamada neu- rofibromina, codificada en el gen del mismo nombre. Al estar el gen mutado la neurofibromina pierde su capacidad supresora tumoral. Se ha asociado la neurofibromatosis con riesgo elevado de cáncer de la vaina nerviosa y con cáncer de colon además de formación de tumores de cualquier naturaleza [1, 2].
Existen básicamente 6 criterios de diagnóstico para la neurofibromatosis:
1) presencia de seis o más manchas de color “café con leche”; 2) dos o más neurofibromas de cualquier tipo o al menos un neurofibroma plexiforme; 3) efélides axilares e inguinales; 4) glioma óptico; 5) dos o más nódulos de Lisch (hamartomas del iris); 6) al menos un familiar en primer grado de consanguinidad que tenga neurofibromatosis tipo 1 [1, 3].
Se trató de una paciente femenina de 32 años de edad, soltera, con instrucción secundaria completa, procedente de Ibarra (provincia de Imbabura-Ecuador) y residente en Quito. Como antecedentes refirió haber tenido desde la infancia manchas de color “café con leche” de predominio en tórax y miembros superiores, así como nódulos subcutáneos. Había sido valorada por varios médicos quienes le indicaron que no era nada patológico. También manifestó que su abuela paterna tenía los mismos signos.
![]()
Su cuadro clínico inició en octubre de 2012, cuando apareció dolor pro- gresivo en la región lumbar con irradiación a miembro inferior izquierdo. Acudió al Hospital Carlos Andrade Marín, del Instituto Ecuatoriano de Seguridad Social, donde fue valorada por Neurocirugía y tuvo como diagnóstico aparente hernia discal. Fue intervenida por esta causa y se halló una masa tumoral, cuyo estudio histopatológico correspondió a un fibroma.
![]()
Revista Médica Vozandes Volumen 24, Número 1-2, 2013
79
![]()
Rodas G
Neurofibroma con alto grado de atipia en una pacien- te con neurofibromatosis tipo 1

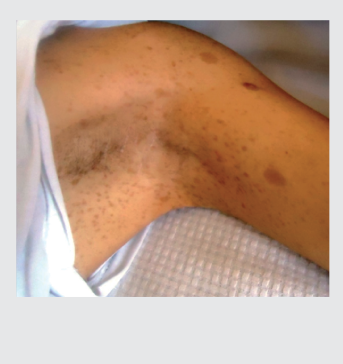
Foto 1. Presencia de manchas “café con leche” y efélides axilares en la paciente.
midiendo entre 1 a 4 cm, de gris blanquecino, aspecto fibroso y áreas friables. Los hallazgos mi- croscópicos mostraron una neoplasia dispuesta en sábanas con células ahusadas de citoplasmas eosinófilos pálidos, con plasmalemas (membranas plasmáticas) no bien definidas, núcleos centrales, grandes, pleomórficos e hipercromáticos, con presencia de alta actividad mitótica; (foto 2). El tumor se acompañaba de extensas zonas de necrosis y los bordes quirúrgicos estuvieron comprometidos. El reporte de inmunohistoquímica indicó CD56 positivo (++ / +++) en células proli- ferantes; vimentina positivo; BCL2 positivo (+++) en células proliferantes; cromogranina negativo; S100 negativo; CD34 positivo en pocas células proliferantes (control interno positivo en vasos); Ki-67 positivo (70%); (foto 3). El diagnóstico fue referido como neurofibroma con atipia e índice de proliferación nuclear alto (70%).
Posteriormente la paciente refirió dolor persistente del miembro inferior izquierdo y notó una tumefacción en el glúteo del mismo lado. Al incrementarse el dolor en intensidad, acudió al Servicio de Emergencias del mis- mo hospital y se le realizó una resonancia magnética del glúteo izquierdo, la cual demostró la presencia de una lesión expansiva, parcialmente encapsulada, con centro necrótico, heterogénea e hiperintensa en las secuencias T2 y STIR, ubicada a nivel de los músculos semitendinoso, aductor mayor y glúteo mayor. Al uso del contraste endo- venoso se observó realce periférico de la lesión y a nivel del músculo cuadrado femoral izquierdo. La lesión medía 13 x 7 x 5 cm aproximadamente.
En la valoración física la paciente se encontraba en condiciones generales regulares y mal estado nutricional. Se identificaron manchas “café con leche” en tórax y miembros superiores; además, efélides axilares bilaterales y un nódulo subcutáneo en antebrazo izquierdo; (foto 1). En tórax se encontró campos pulmonares limpios y murmullo vesicular algo atenuado en bases bilateralmente; ausculta- ción cardíaca normal y glándulas mamarias sin patología aparente. El abdomen fue suave, depresible, algo doloroso en marco colónico; ruidos hidroaéreos de características habituales; región ano genital y perineal normales.
En enero del 2013 se resecó quirúrgicamente la masa, describiéndose como hallazgos operatorios la presencia de un tumor de consistencia heterogénea, originado entre los músculos glúteo mayor y medio, con cápsula dura y un volumen aproximado de 30 cc. El estudio histopatológico reportó a nivel macroscópico múltiples fragmentos de tejido blando irregular, que en conjunto pesaron 88 gramos,
80
La paciente debió ser reintervenida por tres ocasiones debido a recurrencia rápida del tumor. El estudio histopatológico de la última limpieza quirúrgica reportó focos de fibrohistio- sarcoma. La paciente continúa con controles médicos periódicos.
La neurofibromatosis de tipo 1 (NF-1) o enfermedad de Von Recklinghausen, se caracteriza por la formación recurrente y crónica de tumores benignos conocidos como neurofibromas cutá- neos y neurofibromas plexiformes, que son tumores igualmente benignos originados en la vaina de los nervios periféricos, más específicamente en el endoneuro. Su forma de presentación varía de acuerdo a cada familiar afectado, pueden presentarse como nódulos superficiales o subcu- táneos, discretos o de crecimiento difuso y, las complicaciones son impredecibles [1].
![]()
Revista Médica Vozandes Volumen 24, Número 1-2, 2013
Los neurofibromas ocurren en un 25 a 50% de los pacientes afectados por NF-1 y pueden aparecer en fases de crecimiento rápido y alternando con períodos de reposo [1, 3]. Los neurofibromas por lo general están constituidos por células de Schwann, fibroblastos, mastocitos, células del perineuro y axones, todo en una matriz extra- celular. Los neurofibromas plexiformes tienen un aspecto parecido a los subcutáneos pero con una matriz expandida y alta vascularidad periférica y aunque es poco frecuente sufren

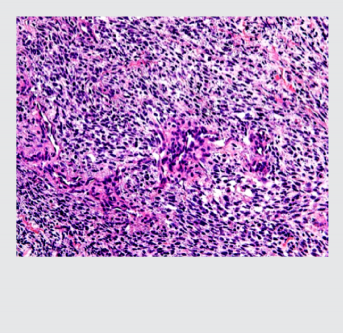
Foto 2. Imagen microscópica de la tumoración. Tinción con hematoxilina-eosina que muestra células en huso de citoplasma eosinófilo.
En cuanto a su diagnóstico, el confirmatorio es el histopatológico y debe ser lo más temprano posible. El tratamiento de elección es la cirugía. El problema es que por su localización (20% columna vertebral, 95% extremidades) no siem- pre la tumoración es resecable. En todo caso, no se ha demostrado ventaja del tratamiento de quimioterapia como primera línea, por lo que se prefiere la radioterapia adyuvante o neo- adyuvante [7].
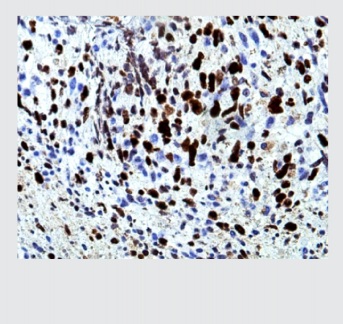
Foto 3. Imagen microscópica de inmunohistoquímica con Ki-67 positivo
El autor declara no poseer conflictos de interés.
transformación maligna [1]. Hasta el 75% de ellos presentan receptores para progesterona [4].
![]()
Revista Médica Vozandes Volumen 24, Número 1-2, 2013
Los pacientes que presentan NF-1 tienen un riesgo de 7% a 10% mayor de padecer un tumor maligno de vaina del nervio periférico [1, 3, 5]. Si comparamos el riesgo de que un neurofibroma plexiforme se transforme en un tumor malig- no, se ha observado que los individuos con NF-1 tienen un 2% a 5% de probabilidades, frente al 0.001% de los que no presentan la enfermedad [1, 6]. Adicionalmente los estudios demuestran que los pacientes afectados con NF-1 y con neurofibromas internos tienen 18 veces más posibilidades de desarrollar un tumor maligno.
Estudio autofinanciado.
El autor es responsable de la síntesis de datos y ela- boración del artículo.
81
Neurofibroma con alto grado de atipia en una pacien- te con neurofibromatosis tipo 1
Rodas G

![]()
Ferner RE, O’Doherty MJ. Neurofibroma and schwannoma. Curr Opin Neurol 2002; 15: 679-84.
Lammert M, Friedman JM, Kluwe L, Maut- ner VF. Prevalence of neurofibromatosis 1 in german children at elementary school enrollment. Arch Dermatol 2005; 141: 71- 74.
Ferner RE. Neurofibromatosis 1 and neuro- fibromatosis 2: a twenty first century pers- pective. Lancet Neurology 2007; 6: 340-51.
3.
4.
5.
McLaughlin ME, Jacks T. Progesterone re- ceptor expression in neurofibromas. Can- cer Res 2003; 63: 752-55.
Tucker T, Wolkenstein P, Revuz J, Zeller J, et al. Association between benign and ma- lignant peripheral nerve sheath tumors in
NF1. Neurology 2005; 65: 205-11.
6.
7.
Ducatman BS, Scheithauer BW, Piepgras DG, Reiman HM, Ilstrup DM. Malignant peripheral nerve sheath tumors. A clinico- pathological study of 120 cases. Cancer 1986; 57: 2006-21.
Baehring JM, Betensky RA, Batchelor TT. Malignant peripheralnerve sheath tumor: the clinical spectrum and outcome of treatment. Neurology 2003; 61: 696-98.
![]()
Revista Médica Vozandes Volumen 24, Número 1-2, 2013
82